以下是生信技能树学员笔记投稿
芯片分析中经常会遇到Affymetrix Human Transcriptome Array 2.0芯片,由于目前还没有现成的R包可以用,因此分析方法也不统一。见生信技能树Jimmy老师HTA2.0芯片比较麻烦,其实这类常见的有3个平台,3种类型:
GPL17586 [HTA-2_0] Affymetrix Human Transcriptome Array 2.0 [transcript (gene) version]GPL19251 [HuGene-2_0-st] Affymetrix Human Gene 2.0 ST Array [probe set (exon) version]GPL16686 [HuGene-2_0-st] Affymetrix Human Gene 2.0 ST Array [transcript (gene) version]
对于这三种平台可以去Affymetrix的官网去查看其区别,也可以去NCBI去查看。
一、获得芯片平台信息文件
通常基因芯片分析,一般要下载其平台信息。一般来说我们下载GPL是为了得到芯片的探针对应基因ID的关系列表。详情可以了解:解读GEO数据存放规律及下载,一文就够一文就够-从GEO数据库下载得到表达矩阵首先是GPL17586平台的芯片,下载其对应的平台文件GPL17586.soft.gz,这类文件通常都比较大,加上国内下载速度太慢,通常都是等了很久,还是下载不了。
查看GPL17586平台下单个的GSE对应的GSEXXX.soft.gz文件,发现其信息与GPL17586.soft.gz相同;下载单个GSE对应的soft文件后,同样可以做id转换。
下面以GPL17586平台下的GSE110359为例,进行id转换,首先是下载GSE110359对应的GSE110359_family.soft.gz文件
1、读入下载好的soft文件
`##
rm(list = ls())
options(stringsAsFactors = F)
#加载R包
library(GEOquery)
gse <- getGEO(filename = "GSE110359_family.soft.gz",destdir = ".")
str(gse)
length(gse)
`
2、提取探针、探针对应的基因及其位于染色体上的位置等信息
`id_probe <- gse@gpls$GPL17586@dataTable@table
dim(id_probe)
head(id_probe)
id_probe[1:4,1:15]
View(head(id_probe))## you need to check this , which column do you need
`
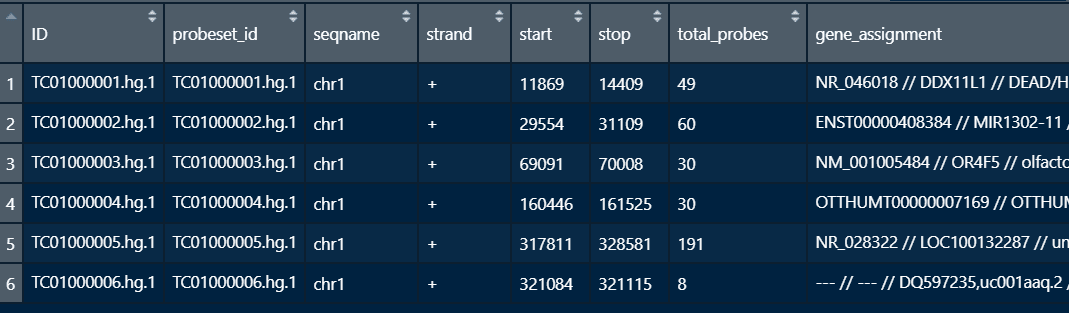
GPL17586平台信息
提取需要的第1列(ID)或者第2列(probeset_id)和第8列(gene_assignment)。当然也可以不提取,每一列都保留。
probe2gene <- id_probe[,c(2,8)]
3、提取第8列 gene_assignment中的基因名称,并添加到probe2gene
`library(stringr)
probe2gene$symbol=trimws(str_split(probe2gene$gene_assignment,'//',simplify = T)[,2])
plot(table(table(probe2gene$symbol)),xlim=c(1,50))
head(probe2gene)
dim(probe2gene)
View(head(probe2gene))
ids2 <- probe2gene[,c(1,3)]
View(head(ids2))
ids2[1:20,1:2]#含有缺失值
table(table(unique(ids2$symbol)))#30907 ,30906个基因,一个空字符
save(ids2,probe2gene,file='gse-probe2gene.Rdata')
`
以上为id转换的第一种方法,下面看第二种方法:
4、使用biomaRt包进行id转换
biomaRt包其实也依赖于网速
`load("gse-probe2gene.Rdata")
dim(probe2gene)
View(head(probe2gene))
加载biomaRt包
library(biomaRt)
value <- probe2gene$probeset_id
attr <- c("affy_hta_2_0","hgnc_symbol")
ensembl <- useMart("ensembl", dataset = "hsapiens_gene_ensembl")
ids <- getBM(attributes = attr,
filters = "affy_hta_2_0",
values = value,
mart = ensembl,
useCache = F)
dim(ids)#[1] 1041 2
View(head(ids))
save(ids,file = "GPL17586_ids.Rdata")
#去重之后
attributes <- listAttributes(ensembl)
View(attributes) # 查看支持的芯片转换格式
save(ids,ensembl,y,file = "ensembl.Rdata")
plot(table(table(ids$hgnc_symbol)),xlim=c(1,50))
table(table(unique(ids$hgnc_symbol)))#去重之后有29262,丢失了一很多
去重复
ids3 <- ids[!duplicated(ids$hgnc_symbol),]
`
综上,可以看出两种方法得到的基因数量差别不大都是从7万多个探针中,获得了差不多3万个基因。
5、表达矩阵和基因id的合并
下面就是表达矩阵和id的合并了;下载表达矩阵,推荐使用Jimmy的万能包GEOmirror一行命令搞定。
`library(GEOmirror)
geoChina(gse = "GSE110359", mirror = "tercent")
library(GEOmirror)
gset <- geoChina(gse = "GSE110359", mirror = "tercent")
gset
a=exprs(gset[[1]])
a[1:4,1:4]
gset[[1]]@annotation
#过滤表达矩阵
exprSet <- a
library(dplyr)
exprSet <- exprSet[rownames(exprSet) %in% ids2$probeset_id,]
dim(exprSet)
exprSet[1:5,1:5]
#ids过滤探针
ids <- ids2[match(rownames(exprSet),ids2$probeset_id),]
dim(ids)
ids[1:5,1:2]
#ids2[1:5,1:2]
#合并表达矩阵和ids
idcombine <- function(exprSet, ids){
tmp <- by(exprSet,
ids$symbol,
function(x) rownames(x)[which.max(rowMeans(x))])
probes <- as.character(tmp)
print(dim(exprSet))
exprSet <- exprSet[rownames(exprSet) %in% probes,]
print(dim(exprSet))
rownames(exprSet) <- ids[match(rownames(exprSet), ids$probeset_id),2]
return(exprSet)
}
new_exprSet <- idcombine(exprSet,ids)
new_exprSet[1:4,1:6]
dim(new_exprSet)
rownames(new_exprSet)
save(new_exprSet,file = "new_exprSet.Rdata")
`
接下来就是质控、差异分析,火山图、GO和KEEG分析,生信技能树上已经有很多这类的推文了,这里就不做演示了。
该方法也适用于GPL16686与GPL19251平台的芯片。只是GPL16686的信息这样的可以用Y叔叔的包进行转换id。
GPL16686平台信息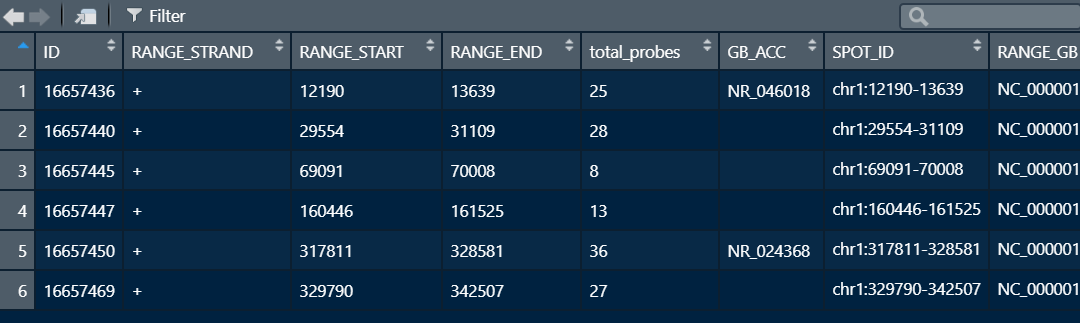
GPL19251平台信息
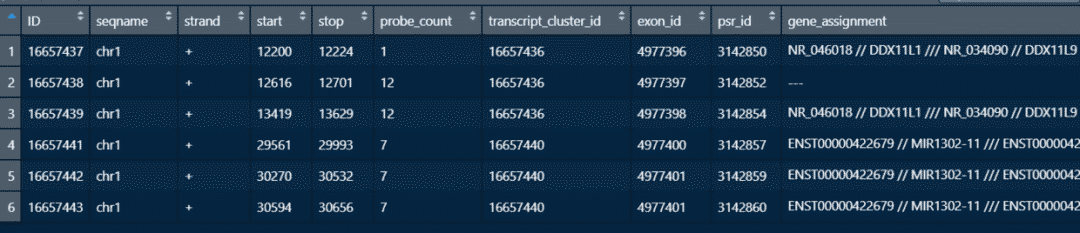
GPL19251平台信息
转换id之后总会有很多探针得不到对应的基因或者很多探针对应一个基因。其实,基因和探针的关系是多对多的关系。
友情推荐:“ID转换和生存分析”群的钉钉群号:35371384,对这几个细节知识点感兴趣的可以加入,我们这个月18号(下周六晚上)八点准时授课。ID转换靠的是深厚的背景知识加上一点代码技巧
写在后面
因为这个投稿是两个星期前的,但是生信技能树学习笔记太多了,所以排班到了今天,上面的ID转换公开课已经结束了,但是今晚有生存分析公开课,感兴趣仍然是可以进去哈,见:生存分析的10年和20年时间点
本文分享自微信公众号 - 生信技能树(biotrainee)。
如有侵权,请联系 support@oschina.cn 删除。
本文参与“OSC源创计划”,欢迎正在阅读的你也加入,一起分享。